General Information


| Student: | Caitlin Guccione |
|---|---|
| Office: | CoRE Building, Rutgers University |
| School: | University of Rhode Island |
| Major: | Mathematics & Computer Science |
| Minor: | Biological Sciences |
| E-mail: | cguccione@my.uri.edu |
| Project: | Elucidating tumor evolutionary patterns using high-depth molecular data |
Project Description
The main goal of this project is to discover the evolutionary patterns of cancer cells so that we can better predict how they develop and in turn resist current treatments. In order to effectively understand how cancer evolves, we must have a strong background in the complexity and patterns within the genomic data. The analysis of cancer patients' genomic data through high-throughput DNA sequencing, along with mathematical modeling, can lead us to a better understanding of cancer tumor growth. We can use trees, a mathematical concept found in graph theory, to model patient data and identify Darwinian selection taking place. Ultimately, we want to create mathematical models of tumor growth to be used on future patients, so that we can better treat cancer before it continues to grow.
Weekly Log
- Week 1:
- My main priorities in week one were to get a better understanding of the background of my project. Reading papers such as The evolution of tumor phylogenetics: principles and practice [1] and Towards Precision Medicine [2], helped me comprehend the biology behind sequencing and other cellular reproduction processes which tie into my project. After reading, I created a presentation describing the background of the project and goals for the summer. Through a paper written by Dr. Khiabanian's lab, I learned about LOHGIC, which is a piece of software proven to be very helpful in the analysis of high-throughput DNA sequencing [3]. More information about LOHGIC and how it works is included below. Finally, I wrapped up the week by learning a bit more about HTML and creating this website.
- Main Accomplishments
- Completed presentation overviewing research for the summer
- Created website
- Goals for next week
- Understand the background on LOHGIC and test it with data
- Learn how to effectively read patient data
- Start doing research on graph theory and trees
- Week 2:
- In week two I focused mainly on learning how the patient data is formatted. There is a large amount of information included with every sample run and not all of it is relevant to tracing the evolution of cancer cells. After picking out the important pieces of data, I learned more about the biology behind them and how accurate these data points were. For example, there is some data that is hard to measure in the field such as purity. Purity measures the amount of the tumor that is cancerous, but there is no good tool to measure this. So, when we look at the sample data, we have to take into account that numbers such as purity may be off. In the future, we are looking to add error bars on purity so that we can make more accurate calculations. In order to make future tasks easier, I also started working on a program that takes patient data files and pulls only the information we need from it. Additionally, I examined the basics of how trees can be used to examine our data.
- Main Accomplishments
- Better understand the patient data files
- Wrote a program to help pull only the information we need from the patient data files
- Goals for next week
- Create a program that given a tumor, out of all the cells with cancer inside the tumor, what percentage have a particular mutation
- Better understand the basic mathematics behind the calculations for our data, for example using the given data to calculate the number of alleles with mutations as opposed to just knowing the purity.
- Week 3:
- I focused on creating a program that given a patient tumor sequencing report, outputs the cancer cell frequency (CCF). This program works by first building off the program I mentioned last week that pulls only the important information from the sequencing report. It then calculates the CCF for if the mutation was Somatic with CNmut=1, Somatic with CNmut =2, loss of heterozygosity(LOH) or Somatic with CNMut=1, LOH. Essentially, we don’t know what type of mutation occurred so we calculate the CCF for all three possible mutations and then see which CCF makes the most sense. Later, we may use LOHGIC to help us filter through these results and pick the mutation that most likely occurred. The other thing I looked into the week was how error propagation works and how this can be built into our calculation for CCF. There is a lot of error on our data, specifically the purity, so error propagation will allow us to have an error bar on our CCF output which leads to more accurate results.
- Main Accomplishments
- Wrote a program that given a patient input file, outputs the cancer cell frequency also known as the CCF
- Learned the basics of Error Propagation
- Goals for next week
- Add error bars to program already written
- Use LOHGIC to help filter CCF results
- Week 4:
- During week four I was finally able to clean up the program I wrote in week three. The first thing I added was error bars. As I mentioned earlier, with so much uncertainty in the data it only makes sense to have a range on the CCF. I added error bars to purity and allele frequency, two data points on the tumor sequencing file. The error bars on purity are simply set at 10% but, the allele frequency error is based of the depth or the number of times that mutation was found in the sample. I also used LOHGIC’s output to filter the data so that only the correct CCF for each data point was created. Finally, I cleaned up the file my program outputs. As for next week, we want to create a master program that calls my initial program multiple times to calculate the differences between tumor samples so we can more easily track cancer evolution.
- Main Accomplishments
- Added error to purity and allele frequency in the program so that the CCF would have error bars as a result
- Updated the program so that it would use LOHGIC’s information to only output the correct CCF
- Goals for next week
- Find the CCF for germline cells, not just somatic ones
- Creating a program that given a patient can track which mutations they had over various tumors across time and how they developed
- Week 5:
- This week, my project took a slight turn. Instead of focusing on using trees to model cancer evolution, we decided to create a program that found the correct purity. The current problem is that multiple purities are given for a sample and they are usually very far off each other and highly inaccurate. While my professor worked on a program that would be able to more accurately track the purity, my job was to create a program called purity_simulation.py that created sample data. This sample data was calculated based on a ‘hidden’ purity kept in another file. This way my professor can test to see if his program finds the correct purity given the sample. Along with that project, I also worked on finding the CCF for germline cells along with somatic ones so if we do go back to modeling, our trees can track both somatic and germline mutation.
- Main Accomplishments
- Calculated the CCF for both germline and somatic cells
- Created a purity simulation program that creates sample data
- Goals for next week
- Test the new purity calculation model to see if it’s accurate
- Incorporate the purity calculation back into the trees and evolution modeling project
- Week 6:
- I focused on updating the programs I had already written this week. The first thing I did was update my GitHub site so that all my code was online. This makes it easier for my professor and graduate students to view my code along with having a backup source of code if something ever happened on my computer. Additionally, I fixed up the program I wrote last week, purity_simulation.py. I added sub-clonal mutations to the simulation since both clonal and sub-clonal mutations occur in real data. I also switched to binomial distribution as opposed to normal distribution when adding noise to the data. This once again follows a more realistic pattern. Finally, I started reading about the best way to represent the error present in our data, onto the trees we are going to build. We have error bars calculated on a majority of it since the sequencing data we use as input is not always accurate. We are currently reading about Nei's distance a paper titled Genetic Distance between Populations [4].
- Main Accomplishments
- Updated Github
- Added Sub-Clonal mutations to purity program so it more accurately represents real data
- Starting reading about how to incorporate error into our trees
- Goals for next week
- Start making trees
- Final presentation
References
-
[1] Schwartz, Russell, and Alejandro A. Schäffer. “The Evolution of Tumour Phylogenetics: Principles and Practice.” Nature News, Nature Publishing Group, 13 Feb. 2017, www.nature.com/articles/nrg.2016.170.
[2] Ashley, Euan A. “Towards Precision Medicine.” Nature News, Nature Publishing Group, 16 Aug. 2016, www.nature.com/articles/nrg.2016.86.
[3] Khiabanian H, Hirshfield KM, Goldfinger M, Bird S, Stein M, Aisner J, Toppmeyer D, Wong S, Chan N, Dhar K, Gheeya J, Vig H, Hadigol M, Pavlick D, Ansari S, Ali S, Xia B, Rodriguez L, Ganesan S. Inference of germline mutational status and evaluation of loss of heterozygosity in high-depth tumor-only sequencing data. JCO Precision Oncology 2018. ASCO.
[4] Nei, M. (1972). Genetic Distance between Populations. The American Naturalist, 106(949), 283-292. Retrieved from http://www.jstor.org/stable/2459777.
Presentations and Papers
- Introduction Presentation
Additional Information
- Mentor
- Dr. Hossien Khiabanian
- Lab Group
- GitHub
Helpful Tools
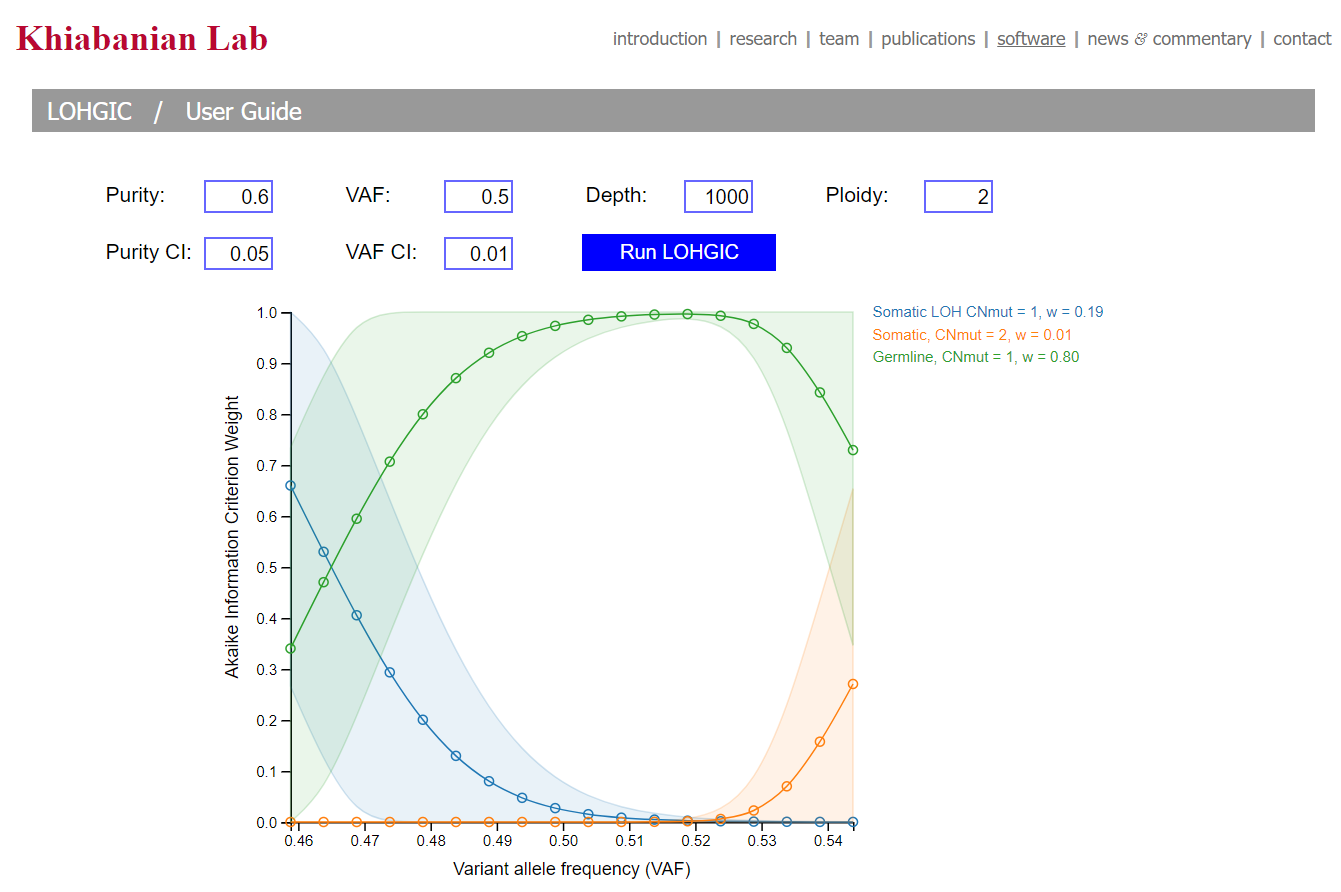
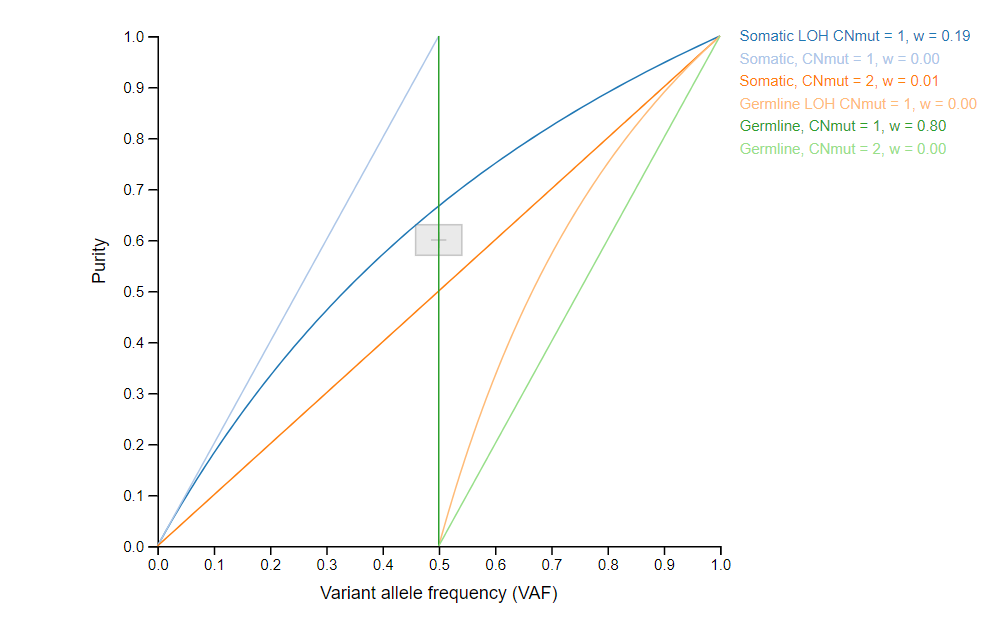
- LOHGIC
- LOHGIC is a helpful tool, produced in Dr.Khiabanian's lab, that takes high-depth DNA sequencing data and determines if mutations are somatic or germline as well as predicting loss of heterozygosity.[3]
-
Try LOHGIC