General Information

| Student: | Ondrej Maxian |
|---|---|
| Office: | Core 446 |
| School: | The Case Western Reserve University |
| E-mail: | oxm60@case.edu |
| Project: | DNA and Chromatin Structures and Folding |
| Mentor: | Wilma Olson |
Project Description
I will be studying nucleosomes this summer. Our DNA comes in a double helix, which usually wraps around proteins called histones to make chromatin. The histone-DNA "bead" is known as a nucleosome, and the packing of nucleosomes controls how our DNA is transcribed. I will be trying to quantify this packing this summer.

What I'm Working On
- Week 1:
- This week I have started to look at nucleosomes. Given two nucleosomes, we can study their overlap
by projecting their polygonal shapes onto a common plane. The steps in this process are:
- Given two nucleosomes, compute their reference frames by computing the eigenvalues and eigenvectors of the deviation matrix from the center (the eigenvectors correspond to the principal axes of the nucleosome), the one with the smallest eigenvalue (smallest deviation from center) is the normal vector because the nucleosome is short and fat (more distance in planar direction than normal).
- Compute a mid-frame and project the two polygonal nucleosomes onto it.
- Compute the overlap of the two polygons
- Week 2:
- This week I filled in some of the details on the list above:
- In week 1, I completed the task of finding the reference frame for 1kx5.
- I used the formulation in [1] to compute the mid-frame of two nucleosomes. This allows us to also find the parameters between the two nucleosomes, specifically shift, slide, rise (translation in the x, y, and z directions of the mid-frame, respectively) and tilt, roll, and twist (rotation in the x, y, and z directions of the mid-frame, respectively).
- After thinking about how to compute the overlap between two polygons, I came up with the following algorithm: given polygons P and Q, first see if there are any points on P inside Q and vice versa. Then add to that list the points where the sides intersect. These points form the overlap polygon, and we can compute its area by breaking it up into triangles and adding.
- Week 3:
- After presenting to the research group last week, this week will be spent refining the model to make sure the results are what they
should be (i.e. is the algorithm actually designed to give us what we are looking for)?
- In some cases, it looks like the midframe approach falls short of giving us the overlap we desire. This seems to happen especially when the nucleosomes are tilted side by side.
- In order to remedy this, we began trying to quantify "side overlap," which is similar to frontal overlap with the exception of the coordinate frame (the vectors essentially "switch" roles here, with the normal vector and one of the other vectors becoming the planar vectors and the other planar vector becoming the normal), and the shape of the polygon (a rectangle instead of an octagon).
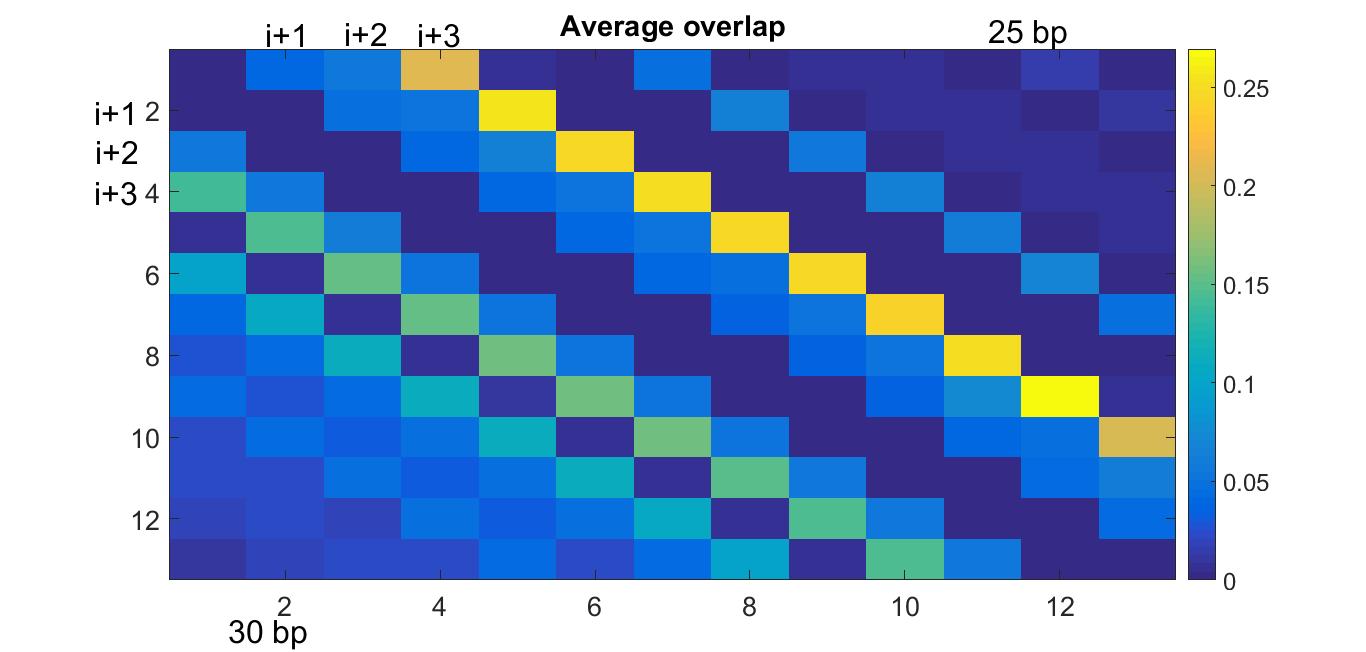
The average overlap matrix for 25 and 30 bp linkers. The (i,j) entry of the matrix is the overlap of nucleosome i with nucleosome j. If j > i, this is for the top half of the matrix, corresponding to 25 bp linkers. The bottom half corresponds to 30 bp linkers.- Week 4:
- This week's focus has been on doing a full analysis of different configurations of nucleosomes. The variable in these configurations is the so-called "linker length" (i.e. the length of the DNA that links the nucleosomes together in base pairs). We expect that more base pairs leads to more flexibility, but we don't know much about how this characterizes the overlap or interaction between the nucleosomes. Some observations from this week:
- Configurations with 25 base pair linker DNA are not as straightforward as we once thought (the original hypothesis was that they displayed a 2 column stacking, meaning there would be a lot of overlap between nucleosome i and i+2. However, we are found out that more of the overlap is from nucleosome i to i+3. This happens because there is a turn of about 140° from one nucleosome to another, which means that nucleosome i falls between i+2 and i+3.
- Configurations with 15 base pairs, 18 base pairs (pictured below), and 20 base pairs show vastly different overlap behavior because of the degree of twisting that they exhibit. For example, 18 base pair linkers show an almost perfect 120° twist, which means that they overlap with nucleosomes i+3 and i+6 (see photo and video below). 15 base pairs twists a little less than this and 20 is a little more, which leads to a little more confusing overlap matrices.
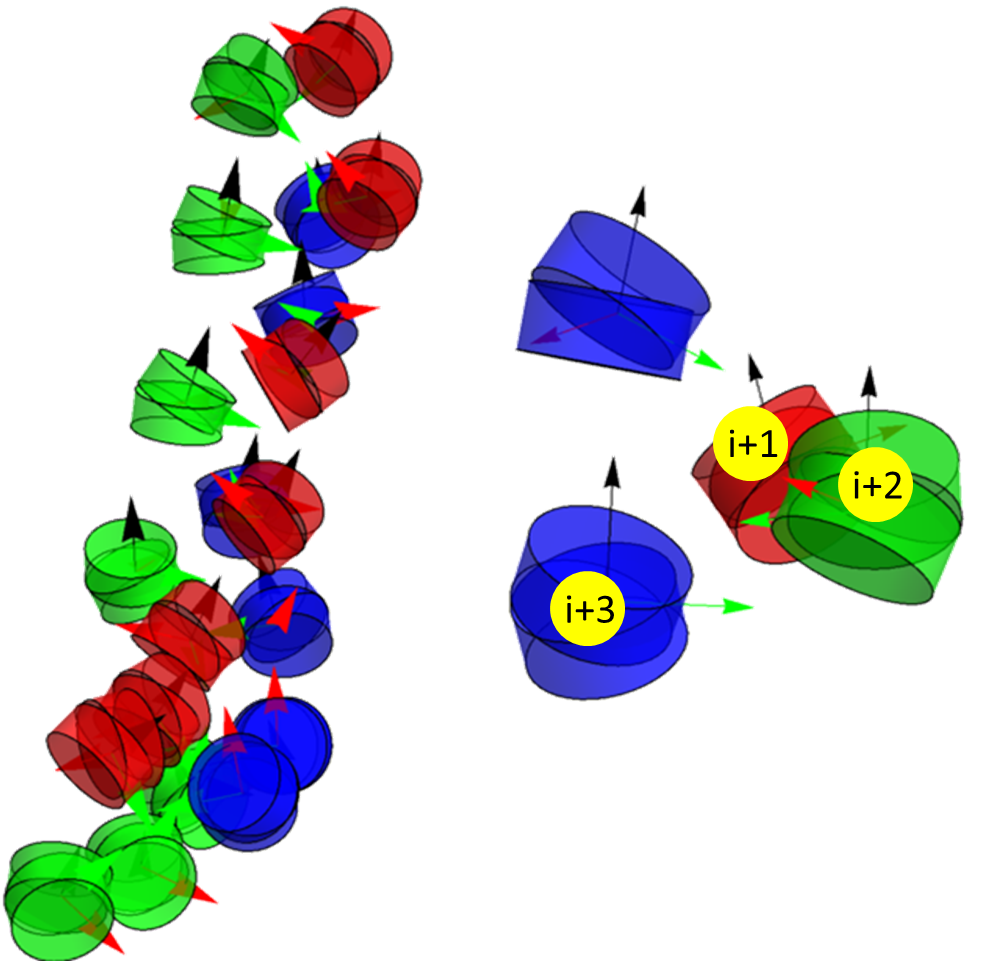
18 base pair configurations (with 20 nucleosomes) that show i+3 overlap
A video of the overlap matrix in 18 bp linkers evolving over time. The lower triangle shows the average over all of the processed configurations, while the upper half shows the overlap matrix of that particular configuration. Note the strong overlap in the i+3 and i+6 directions, as suggested by the configuration shown.- Week 5:
- This week the focus has been on analyzing nucleosome configurations under two situations. The first is when a linker histone protein is added to the apparatus. It seems that in cases when we fix the 9 base pairs on the exit of the nucleosome (as an assumed response to the histone protein), winding is facilitated. Meanwhile, when we don't fix the exit base pairs (i.e. when we fix the entry or when the histone is free), there is no winding and stacking results instead (in this case, there is more overlap as you go down a column, see picture below). This trend held for 40 base pair and 30 base pair linker DNA. 50 and 60 base pair linkers didn't show this trend, as they tended to be more separated, have less overlap, and interact with each other less.
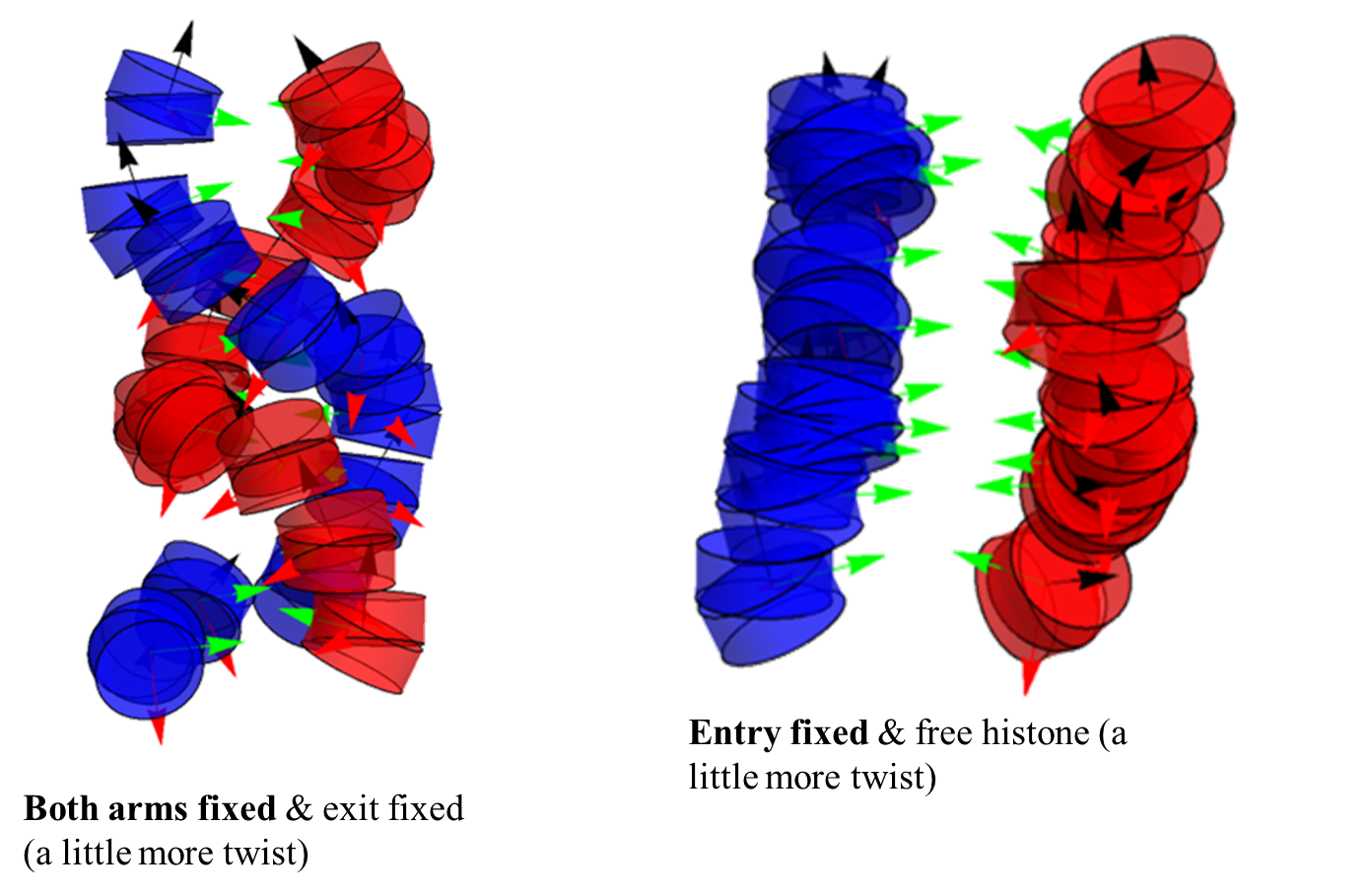
Configurations with a linker histone protein and 30 bp linkers. The boldface type indicates the configuration shown, and the normal type shows the other kind of configuration that looks similar (except the other configuration has a little more twist).
I have also started looking at what happens when we remove the histone tails. Within the nucleosome, the core histone proteins have tails that stick out of the nucleosome shape (you can see them protruding out from the picture at the top of the page). The hypothesis is that these tails create more winding of the nucleosomes in a configuration. We see that in some cases this is true, as configurations with tails have less overlap than those with no tails. Less overlap means that the nucleosomes are not as "stacked" on top of each other, meaning there is more twisting. We verified this by looking at an end to end distance plot of the configurations, finding that on average the two ends were closer with tails than without.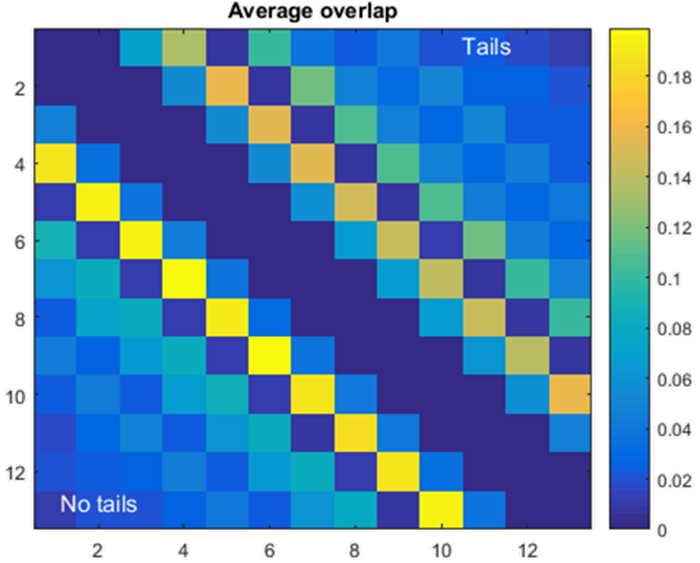
Comparison of overlaps between nucleosomes i and j for configurations with histone tails (top half of matrix, entries where column > row [j > i]) and without tails (bottom half of matrix, where column < row [j < i]). Notice that configurations with tails have less overlap, likely because they twist more.- Week 6:
- This week has been short because of the Independence Day Holiday. However, I still made some progress looking at the tailless nucleosomes in more detail. The diagram below shows a nucleosome with all 8 of the tails labeled. Note that there are 2 tails of each of the 4 proteins (H2A, H2B, H3, H4). We found from the simulation results that removing the H2AH2B tails has no effect, whereas removing the H3H4 tails increases overlap and reduces twisting.
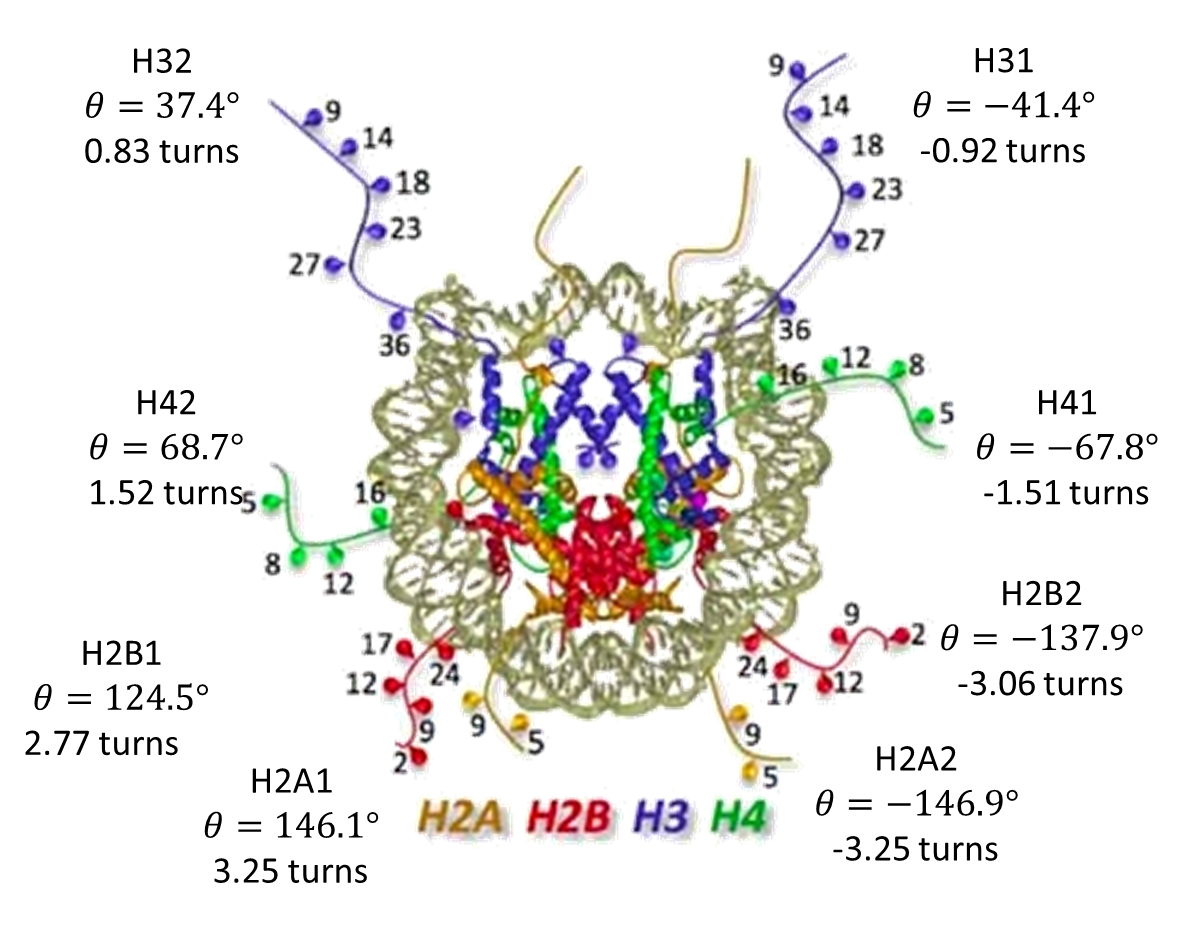
The locations of the tails. The θ angle goes from -180° to 180° and shows the twist from the dyad where the tail is located if projected onto the center plane. The turns parameter is the number of helical turns.
Now, we examined a distribution of where the nucleosomes in 30 bp linker DNA chromatin overlap each other from the side (as a function of the number of helical turns away from the dyad) and compared it to the locations of the tails. The photo below shows that in 30 bp DNA, the nucleosomes contact each other around the H3 tails. This is why the H3 tails make such a difference, because they are being added where the nucleosomes are facing each other!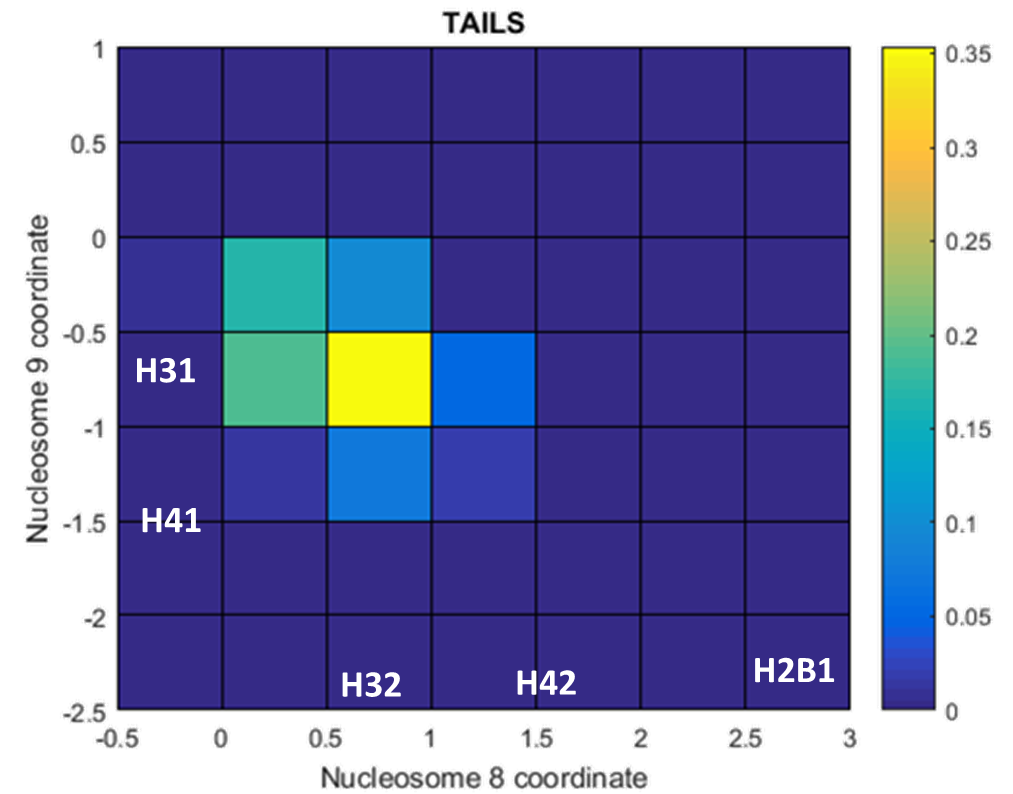
Distribution of where nucleosomes in 30 bp linker DNA chromatin face each other from the sides. Observe that most of the side overlap occurs from 0.5 to 1 helical turn in nucleosome i and from -0.5 to -1 helical turn in nucleosome i+1. These correspond to the H32 and H31 tails, respectively.- Week 7:
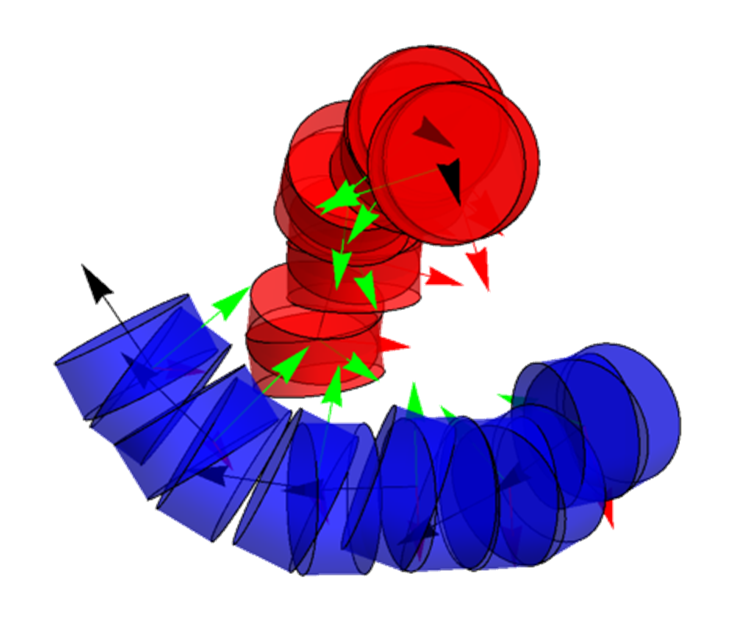
- This week I have started to look at crystal structures, which happen when nucleosomes are packed very tightly (see sample below).
A crystal structure
Biological experiments tell us that in crystals, the acidic patch (negatively charged) on the H2A/H2B dimer interacts with the positive H42 tail on the other nucleosome (see picture from last week). We tried to confirm this by computing the side overlap along with its direction, and then looking at the proportion of contacts that were in the acidic patch to H42 tail direction. The picture below shows the frequency of acidic patch to H42 tail contact as a function of the amount of overlap. One can see that when overlap is high, there is almost always H42-acidic patch contact.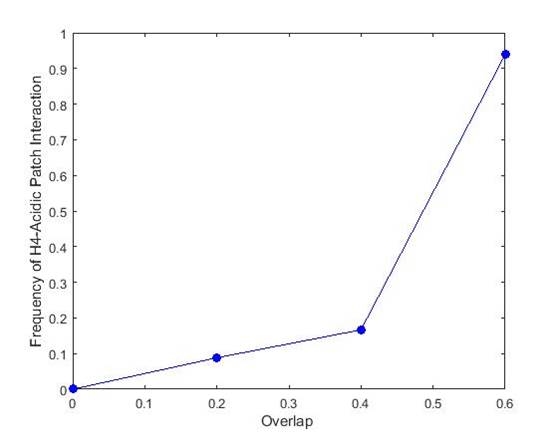
The frequency of acidic patch to H42 tail contact as a function of the amount of overlap between the 2 nucleosomes.
Now, the overlap is almost always H42 to acidic patch, but there are some cases when part of the H2A1 core histone interacts with the H32 and H42 core histones in different locations. (a shift of about 40°, if you consider the cylinder base as a circle in polar coordinates). We are continuing to try to understand why this happens and how it affects the crystal structure.- Week 8:
- This week I have begun consolidating my results for potential publication. My goal will be to show the differences and similarities in nucleosome interactions in crystals, fibers from cryogenic electron microscopy (cryo-EM for short, like one shown below), and simulations of some fixed linker length.
A cryo EM fiber structure.
In all cases, we are finding that nucleosomes that have the largest overlap between them tend to be in contact via the H4-acidic patch interaction. Unlike the crystal structures, where this tends to be obvious for high overlaps, the cryo-EM structure doesn't show this as clearly, and the simulations tend to be more ambiguous. The heatmap below shows the average (x,y) coordinate of interaction on the top face of a nucleosome, with the centers of the core histones plotted on top of it. Note the region of most interaction is between H4 and the acidic patch, which is remarkable given the flexibility allotted to the simulations.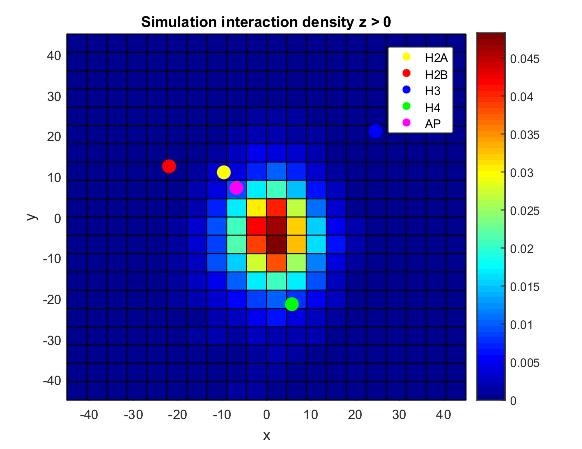
The regions of contact on the top face of the nucleosomes in the simulations. Note that the region is between the acidic patch (AP) and H4.- Week 9:
- This week I have been working on my final technical report as well as code documentation so that others can use the software I developed. Thanks to everyone who made this a great REU!
Presentations
References
- Lu et al. Structure and Conformation of Helical Nucleic Acids: Analysis Program (SCHNAaP). J. Mol Bio (1997) 273, 668-680.